Click-Free Synthesis of a Multivalent Tricyclic Peptide as a Molecular Transporter

 and
and
Abstract
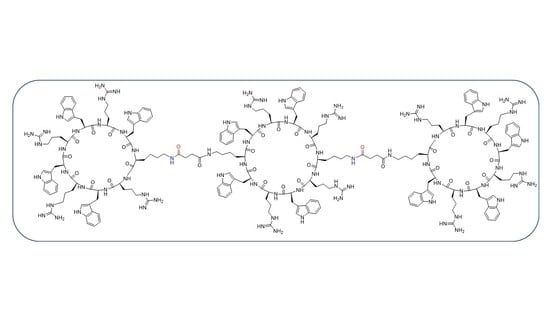
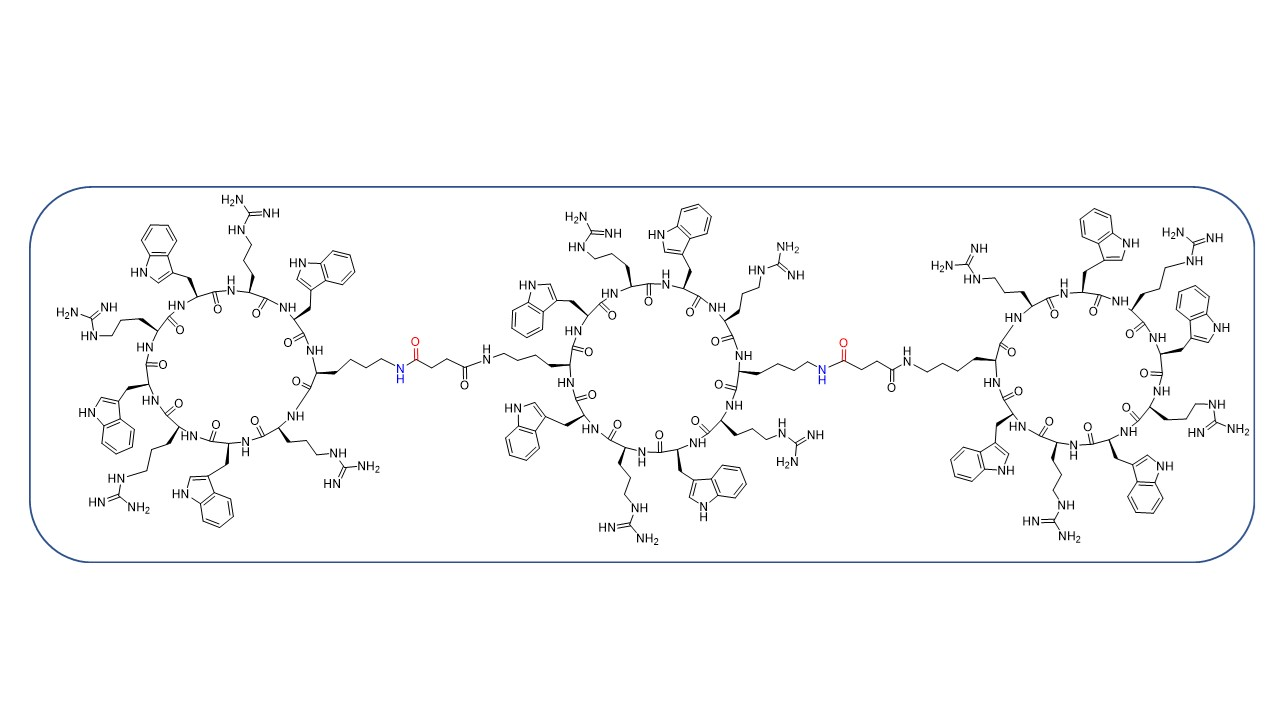
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chemistry
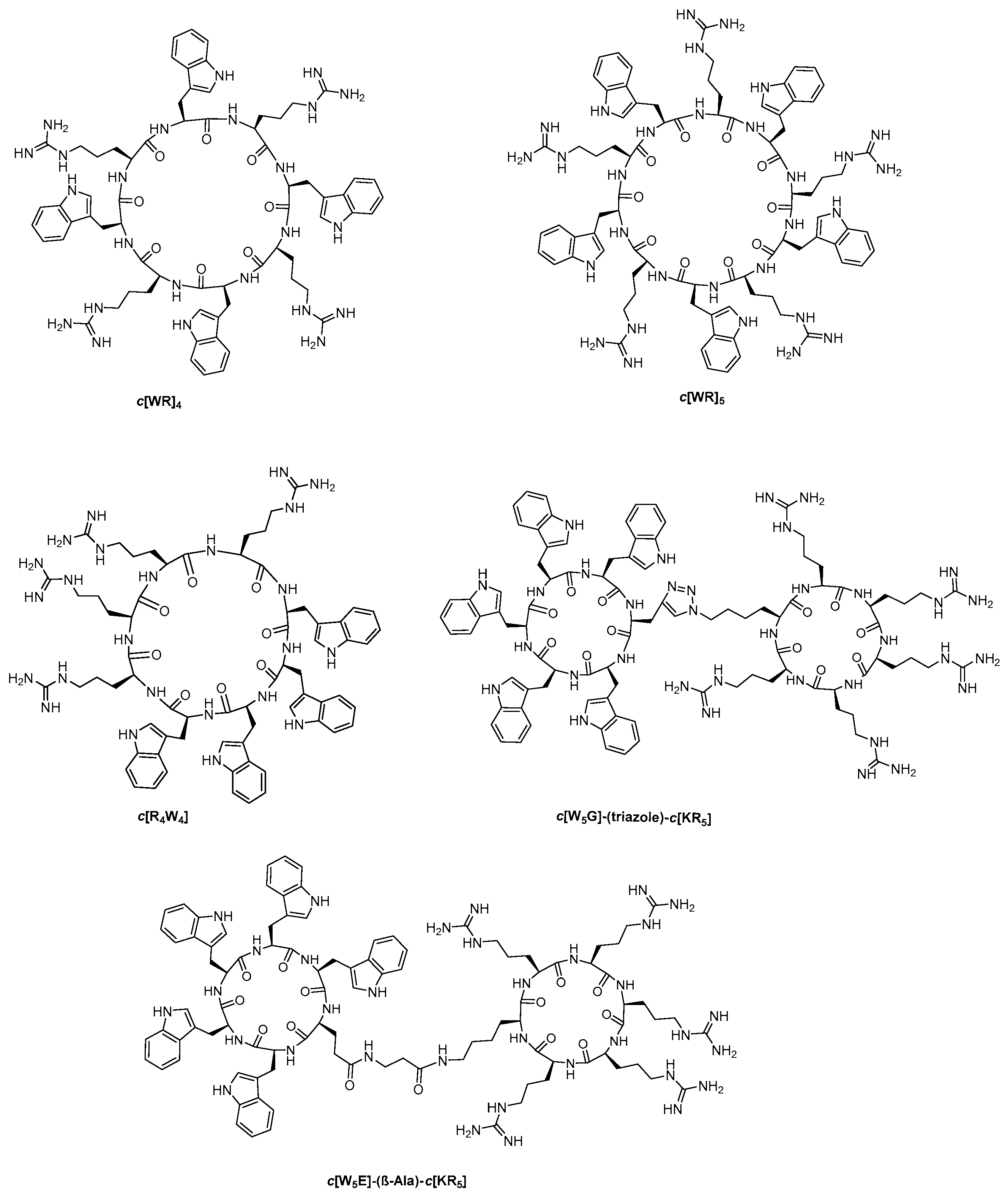
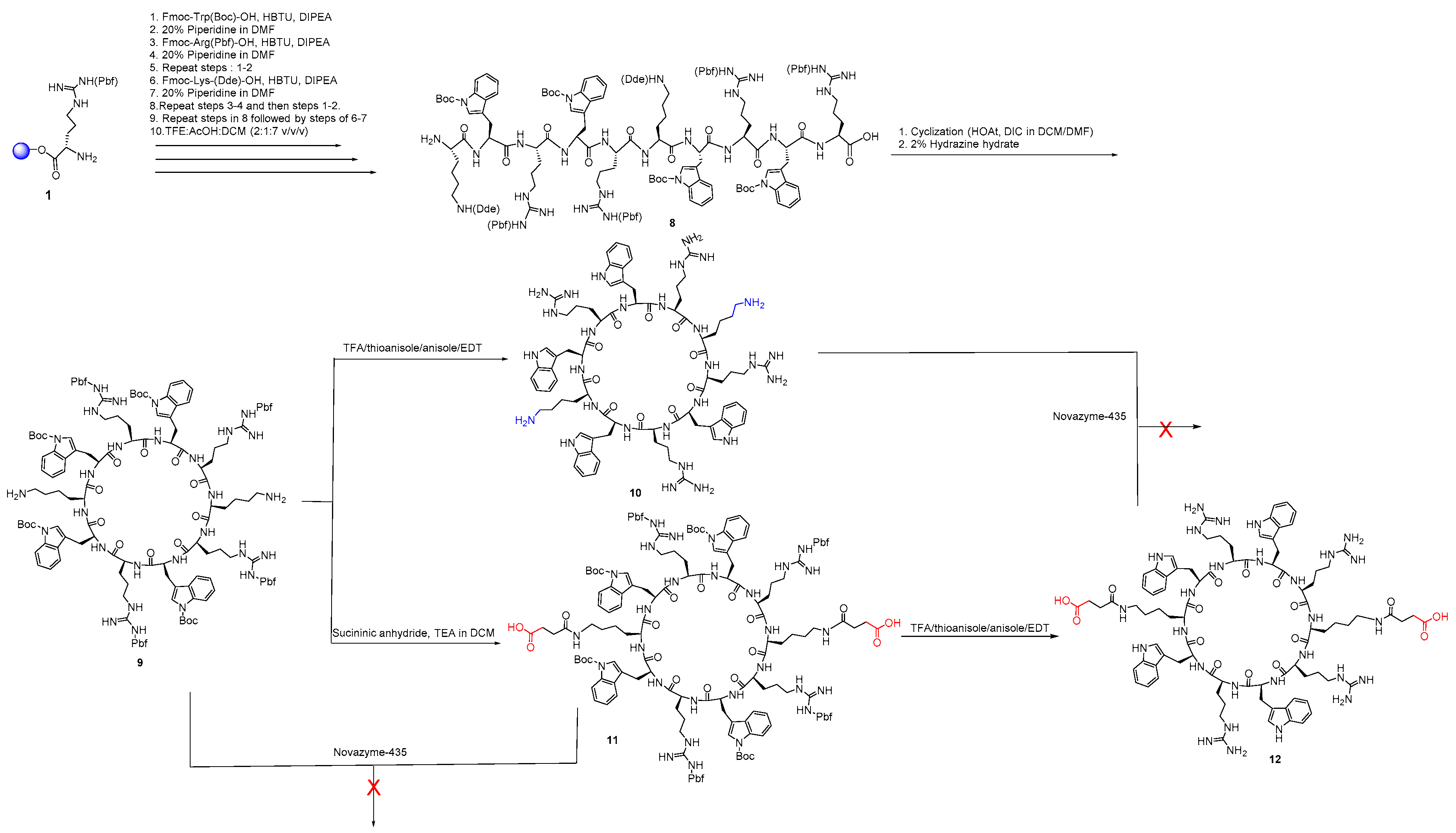
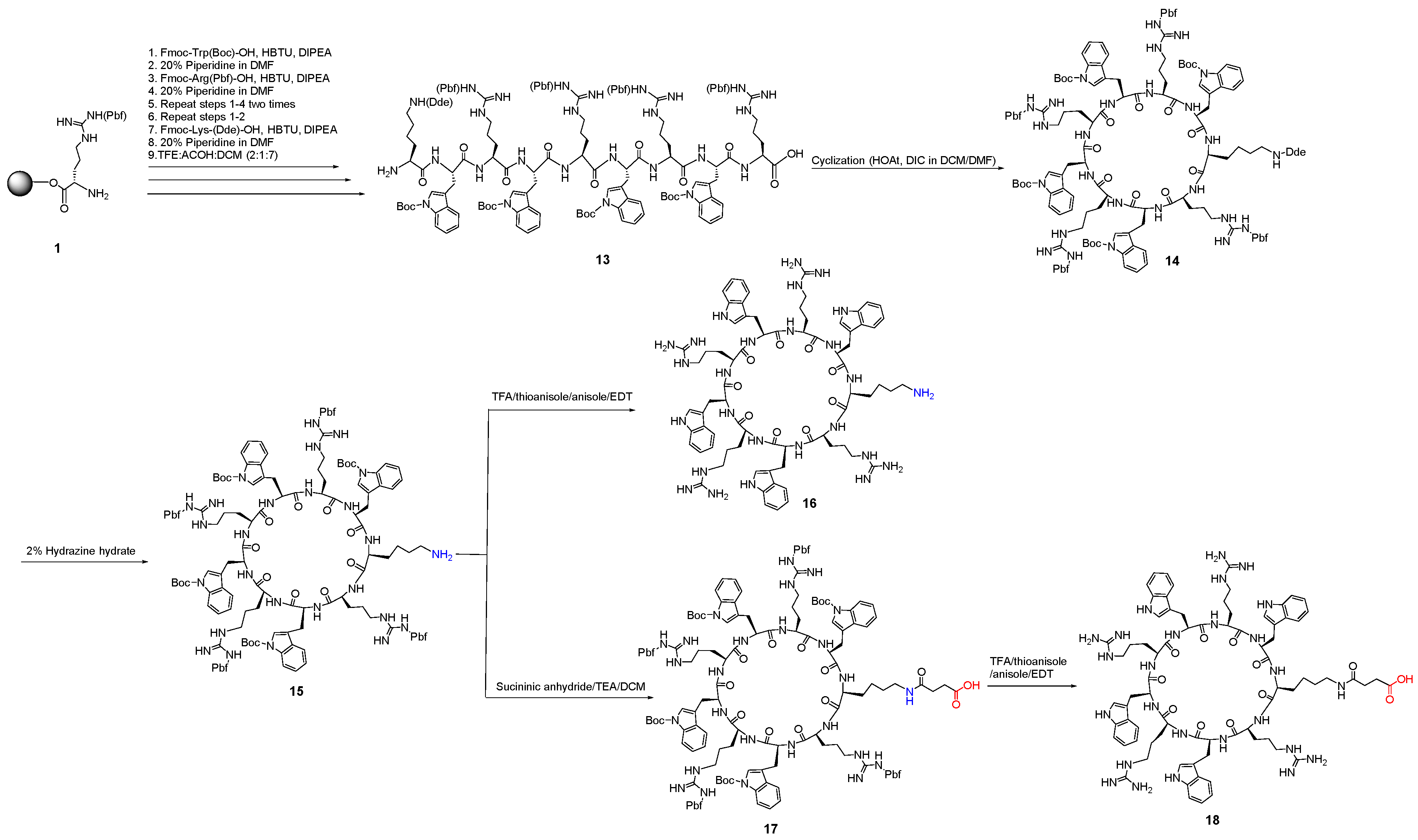
2.2.1. General Method for the Synthesis of Monocyclic Peptides
2.2.2. Synthesis of Monocyclic Cyclic Peptides Containing Free Amino or Free Carboxylic Acid or Both
2.2.3. Synthesis of Diamine Cyclic Peptide [KWRWRKWRWR] (10) and Diacid Cyclic Peptide [K(COCH2CH2COOH)WRWRK(COCH2CH2COOH)WRWR] (12)
2.2.4. Synthesis of a Monoamine Cyclic Peptide [RWRWKRWRW] (16) and a Mono Carboxylic Acid Cyclic Peptide [K(COCH2CH2COOH)-WRWRWRWR] (18)
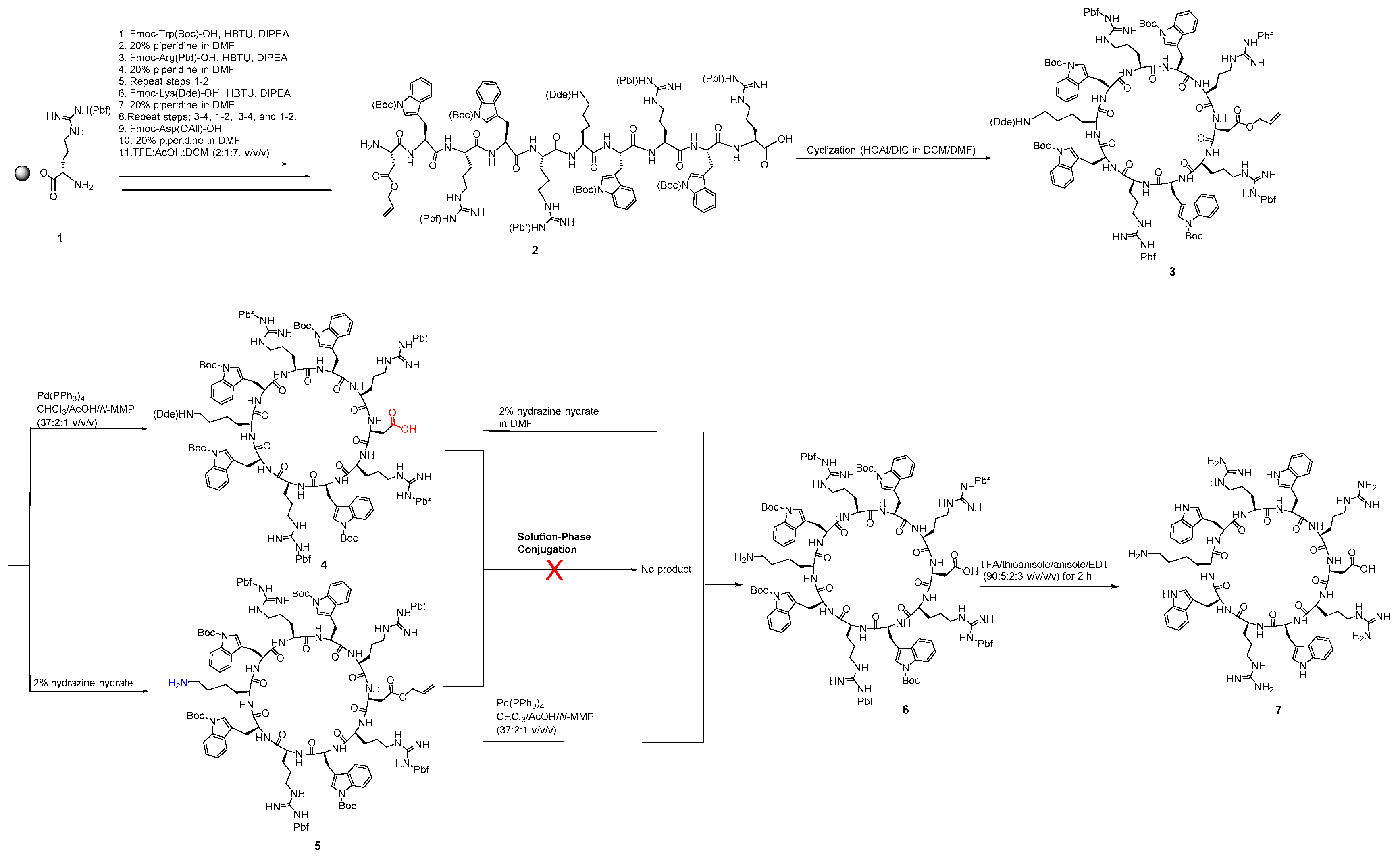
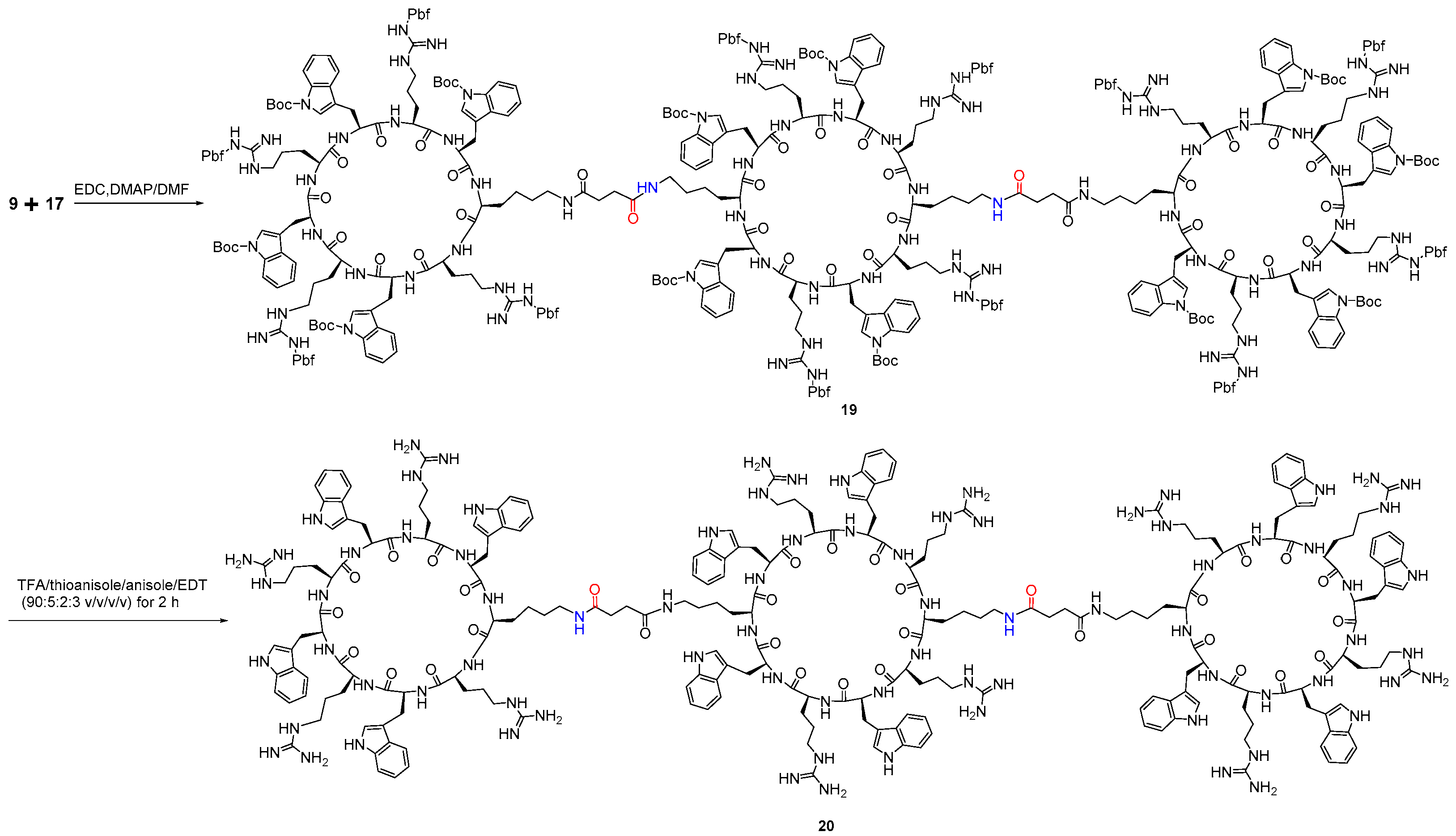
2.2.5. Synthesis of Tricyclic Peptide 20
2.3. Bioassay
2.3.1. Antibacterial Assay
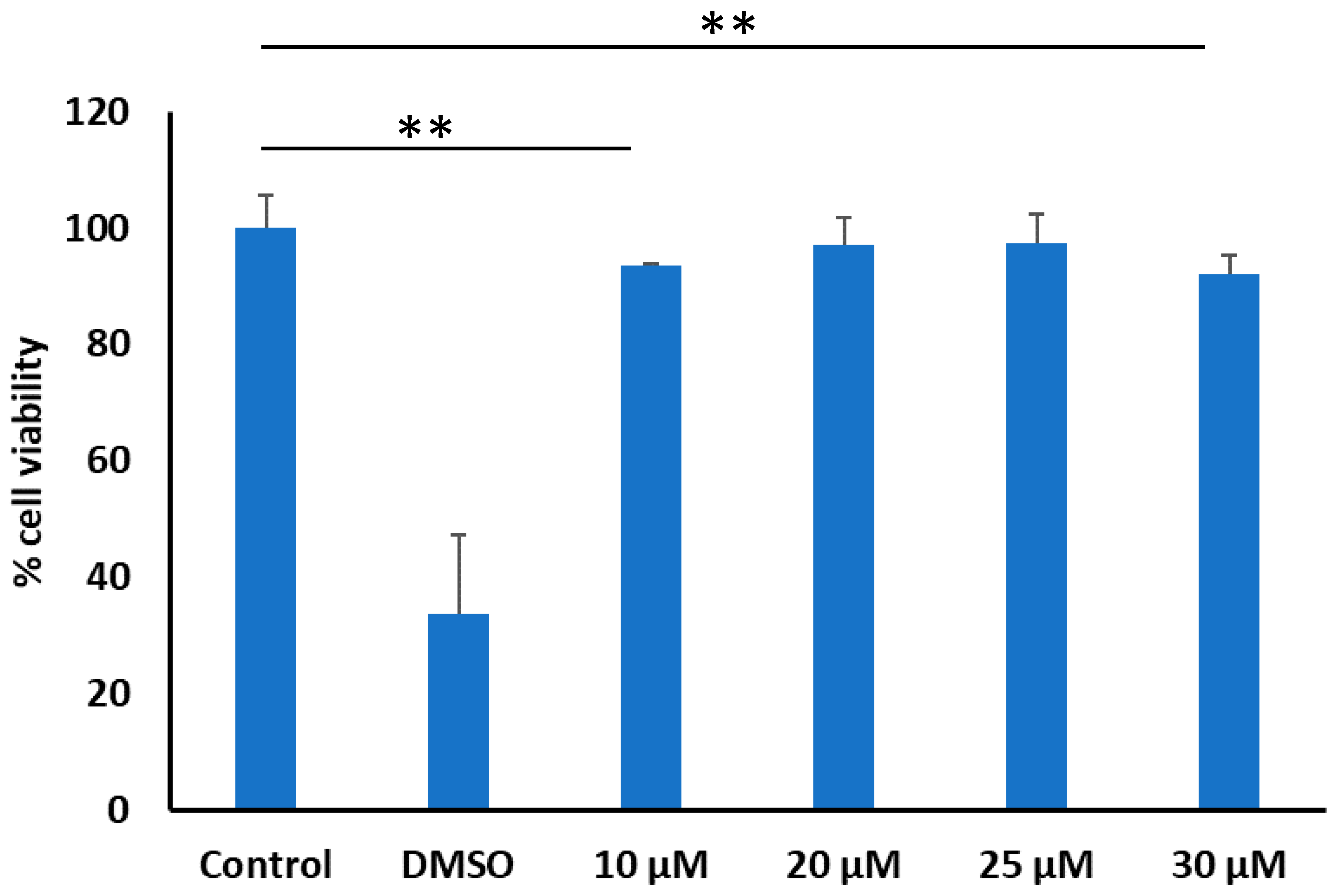
2.3.2. Cellular Cytotoxicity Assay
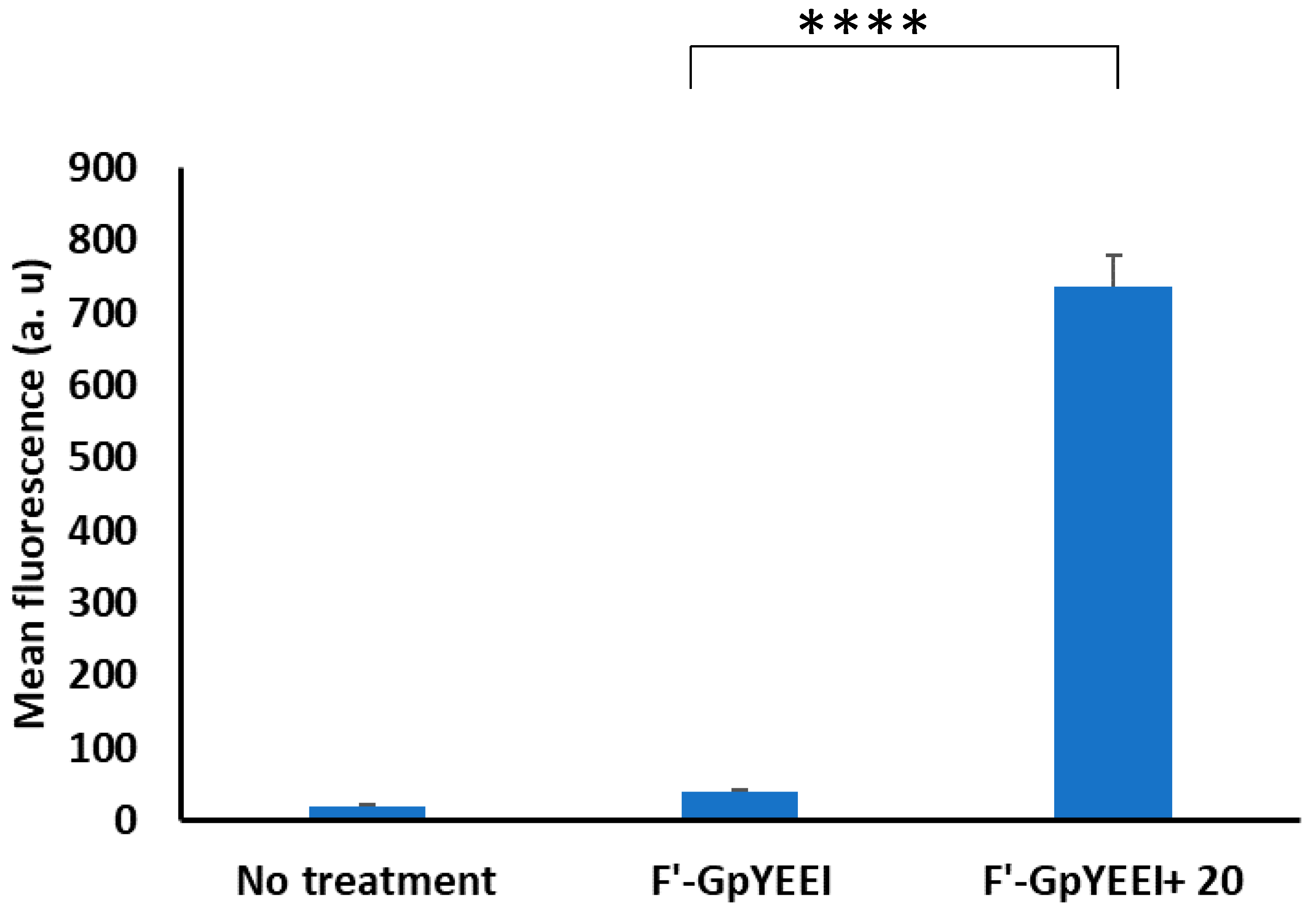
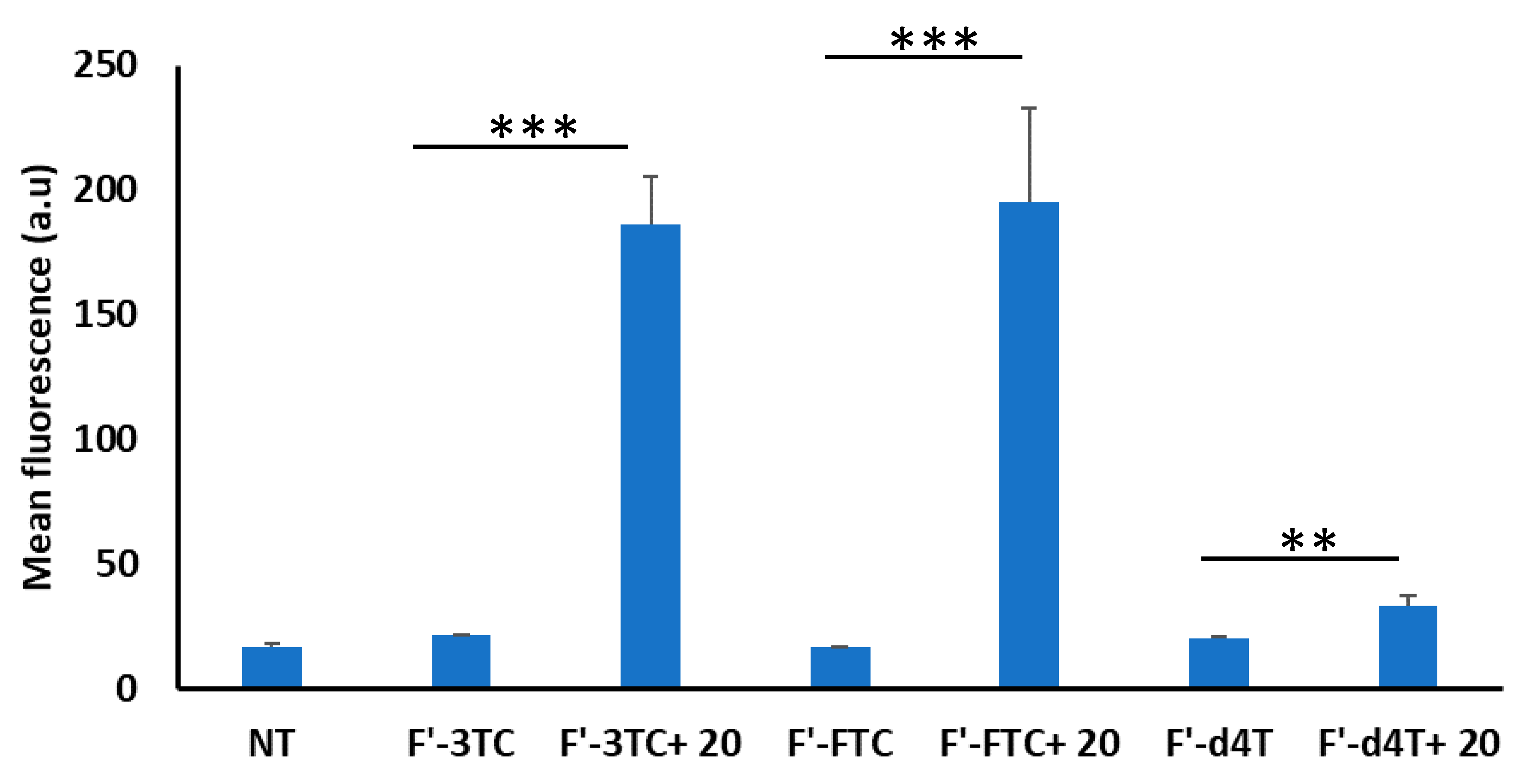
2.3.3. Cellular Uptake of Fluorescence-Labeled Phosphopeptide (F’-GpYEEI) and Anti-HIV Drugs (Lamivudine (F’-3TC), Emtricitabine (F’-FTC), and Stavudine (F’-d4T)
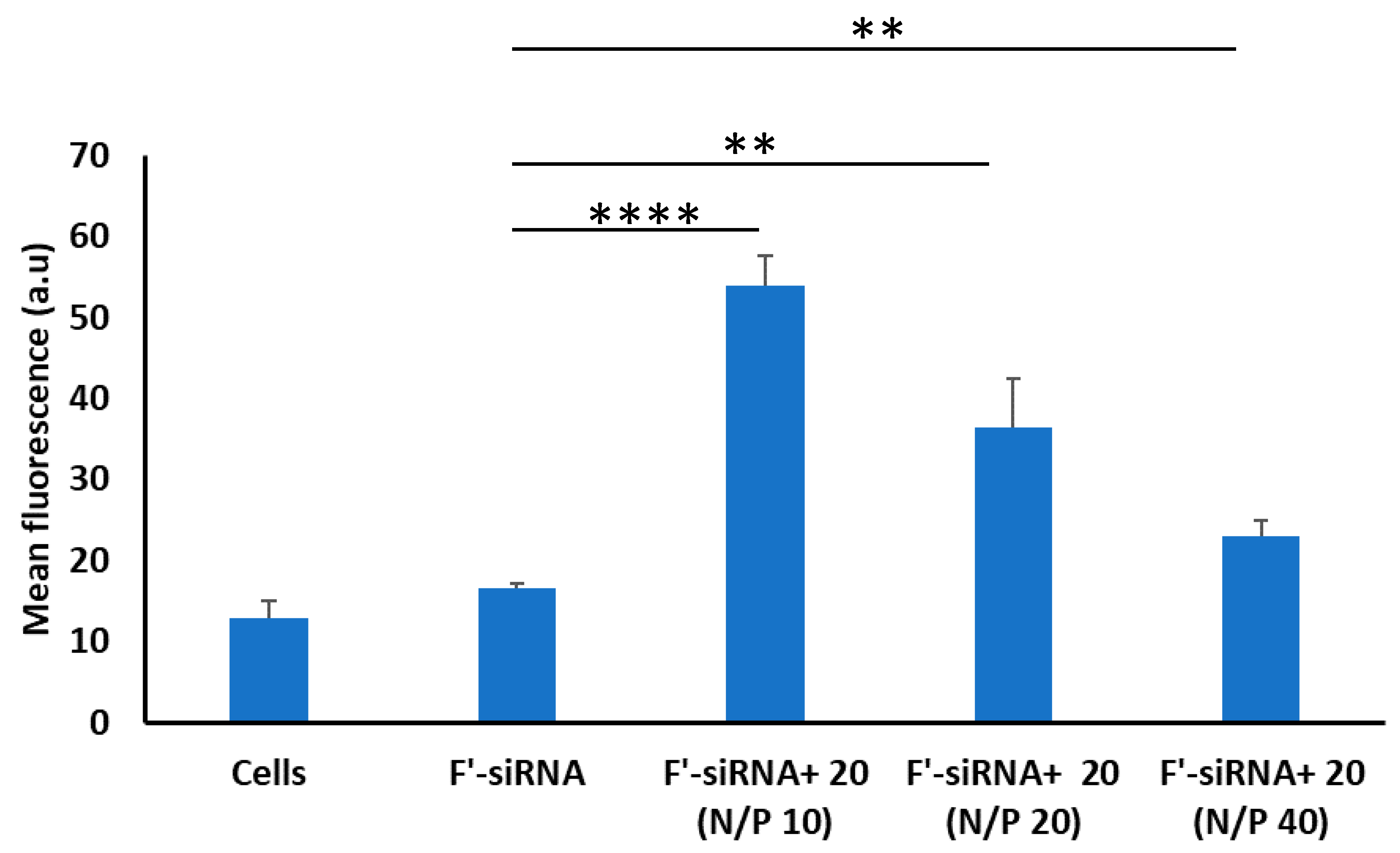
2.3.4. siRNA Delivery
2.3.5. Statistical Analyses
3. Results & Discussion
3.1. Chemistry
3.2. Biological Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent aptamers: Versatile tools for diagnostic and therapeutic applications. Molecules 2016, 21, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compain, P. Multivalent effect in glycosidase inhibition: The end of the beginning. Chem. Rec. 2020, 20, 10–22. [Google Scholar] [CrossRef]
- Long, D.D.; Aggen, J.B.; Christensen, B.G.; Judice, J.K.; Hegde, S.S.; Kaniga, K.; Krause, K.M.; Linsell, M.S.; Moran, E.J.; Pace, J.L. A multivalent approach to drug discovery for novel antibiotics. J. Antibiot. (Tokyo) 2008, 61, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhan, P.; Li, X.; Rai, D.; De Clercq, E.; Liu, X. Multivalent agents: A novel concept and preliminary practice in Anti-HIV drug discovery. Curr. Med. Chem. 2013, 20, 815–832. [Google Scholar] [PubMed]
- Haag, R. Multivalency as a chemical organization and action principle. Beilstein J. Org. Chem. 2015, 11, 848–849. [Google Scholar] [CrossRef] [Green Version]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. Engl. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [Green Version]
- Satav, T. The self-assembly and dynamics of weakly multivalent, peptide-based, host guest systems. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2015. [Google Scholar]
- Zacco, E.; Anish, C.; Martin, C.E.; Hans, V.B.; Brandenburg, E.; Seeberger, P.H.; Koksch, B. A Self-assembling peptide scaffold for the multivalent presentation of antigens. Biomacromolecules 2015, 16, 2188–2197. [Google Scholar] [CrossRef] [Green Version]
- Henning, L.M.; Bhatia, S.; Bertazzon, M.; Marczynke, M.; Seitz, O.; Volkmer, R.; Haag, R.; Freund, C. Exploring monovalent and multivalent peptides for the inhibition of FBP21-tWW. Beilstein J. Org. Chem 2015, 11, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Lauster, D.; Glanz, M.; Bardua, M.; Ludwig, K.; Hellmund, M.; Hoffmann, U.; Hamann, A.; Bottcher, C.; Haag, R.; Hackenberger, C.P.R.; et al. Multivalent peptide-nanoparticle conjugates for influenza-virus inhibition. Angew. Chem. Int. Ed. Engl. 2017, 56, 5931–5936. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. (Seoul) 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gang, D.; Kim, D.W.; Park, H.S. Cyclic Peptides: Promising saffolds for biopharmaceuticals. Genes 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, S.; Koyasu, S. Mechanisms of action of cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding cell penetration of cyclic peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed. Engl. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- El-Sayed, N.S.; Shirazi, A.N.; Sajid, M.I.; Park, S.E.; Parang, K.; Tiwari, R.K. Synthesis and antiproliferative activities of conjugates of paclitaxel and camptothecin with a cyclic cell-penetrating peptide. Molecules 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, A.N.; Paquin, K.L.; Howlett, N.G.; Mandal, D.; Parang, K. Cyclic peptide-capped gold nanoparticles for enhanced siRNA delivery. Molecules 2014, 19, 13319–13331. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Tiwari, R.K.; Oh, D.; Banerjee, A.; Yadav, A.; Parang, K. Efficient delivery of cell impermeable phosphopeptides by a cyclic peptide amphiphile containing tryptophan and arginine. Mol. Pharm. 2013, 10, 2008–2020. [Google Scholar] [CrossRef] [Green Version]
- Nasrolahi Shirazi, A.; Tiwari, R.K.; Oh, D.; Sullivan, B.; Kumar, A.; Beni, Y.A.; Parang, K. Cyclic peptide-selenium nanoparticles as drug transporters. Mol. Pharm. 2014, 11, 3631–3641. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499. [Google Scholar] [CrossRef]
- Oh, D.; Darwish, S.A.; Shirazi, A.N.; Tiwari, R.K.; Parang, K. Amphiphilic bicyclic peptides as cellular delivery agents. ChemMedChem 2014, 9, 2449–2453. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.; Sun, J.; Nasrolahi Shirazi, A.; LaPlante, K.L.; Rowley, D.C.; Parang, K. Antibacterial activities of amphiphilic cyclic cell-penetrating peptides against multidrug-resistant pathogens. Mol. Pharm. 2014, 11, 3528–3536. [Google Scholar] [CrossRef] [PubMed]
- Riahifard, N.; Mozaffari, S.; Aldakhil, T.; Nunez, F.; Alshammari, Q.; Alshammari, S.; Yamaki, J.; Parang, K.; Tiwari, R.K. Design, synthesis, and evaluation of amphiphilic cyclic and linear peptides composed of hydrophobic and positively-charged amino acids as antibacterial agents. Molecules 2018, 23, 2722. [Google Scholar] [CrossRef] [Green Version]
- Mahon, E.; Barboiu, M. Synthetic multivalency for biological applications. Org. Biomol. Chem. 2015, 13, 10590–10599. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, L.L.; Gestwicki, J.E.; Strong, L.E. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr. Opin. Chem. Biol. 2000, 4, 696–703. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef]
- Lee, J.; Bai, Y.; Chembazhi, U.V.; Peng, S.; Yum, K.; Luu, L.M.; Hagler, L.D.; Serrano, J.F.; Chan, H.Y.E.; Kalsotra, A.; et al. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2019, 116, 8709–8714. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Soares, E.V.; Hebbelinck, K.; Soares, H.M. Toxic effects caused by heavy metals in the yeast Saccharomyces cerevisiae: A comparative study. Can. J. Microbiol. 2003, 49, 336–343. [Google Scholar] [CrossRef]
- Brewer, G.J. Risks of copper and iron toxicity during aging in humans. Chem. Res. Toxicol. 2010, 23, 319–326. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.A.; Klok, H.A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. (Camb.) 2008, 2591–2611. [Google Scholar] [CrossRef]
- Jiang, Y.; Maniar, D.; Woortmana, A.J.J.; Loos, K. Enzymatic synthesis of 2,5-furandicarboxylic acid-based semi-aromatic polyamides: Enzymatic polymerization kinetics, effect of diamine chain length and thermal properties. RSC Adv. 2016, 6, 67941–67953. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.N. Enzyme-catalyzed synthesis of polyamides and polypeptides. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA.: Weinheim, Germany, 2010; pp. 131–141. [Google Scholar]
- Zhou, Y.; Abagyan, R. How and why phosphotyrosine-containing peptides bind to the SH2 and PTB domains. Fold. Des. 1998, 3, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Machida, K.; Mayer, B.J. The SH2 domain: Versatile signaling module and pharmaceutical target. Biochim. Biophys. Acta 2005, 1747, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Steinstraesser, L.; Kraneburg, U.M.; Hirsch, T.; Kesting, M.; Steinau, H.U.; Jacobsen, F.; Al-Benna, S. Host defense peptides as effector molecules of the innate immune response: A sledgehammer for drug resistance? Int. J. Mol. Sci. 2009, 10, 3951–3970. [Google Scholar] [CrossRef] [Green Version]
- Niyonsaba, F.; Nagaoka, I.; Ogawa, H.; Okumura, K. Multifunctional antimicrobial proteins and peptides: Natural activators of immune systems. Curr. Pharm. Des. 2009, 15, 2393–2413. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of human defensin mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | MIC (µg/mL) # | |||
|---|---|---|---|---|
| MRSA (LAC Clone) | Klebsiella pneumoniae (ATCC BAA 1705) | Pseudomonas aeruginosa (ATCC 27883) | E. coli ATCC 25922) | |
| Meropenem | 4 | 32 | 1 | 8 |
| c[R4W4] | 4 | 32 | 64 | 16 |
| Tricyclic Peptide 20 | 64 | 128 | 128 | 64 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Mandal, D.; El-Mowafi, S.A.; Mozaffari, S.; Tiwari, R.K.; Parang, K. Click-Free Synthesis of a Multivalent Tricyclic Peptide as a Molecular Transporter. Pharmaceutics 2020, 12, 842. https://doi.org/10.3390/pharmaceutics12090842
Kumar S, Mandal D, El-Mowafi SA, Mozaffari S, Tiwari RK, Parang K. Click-Free Synthesis of a Multivalent Tricyclic Peptide as a Molecular Transporter. Pharmaceutics. 2020; 12(9):842. https://doi.org/10.3390/pharmaceutics12090842
Chicago/Turabian StyleKumar, Sumit, Dindyal Mandal, Shaima Ahmed El-Mowafi, Saghar Mozaffari, Rakesh Kumar Tiwari, and Keykavous Parang. 2020. "Click-Free Synthesis of a Multivalent Tricyclic Peptide as a Molecular Transporter" Pharmaceutics 12, no. 9: 842. https://doi.org/10.3390/pharmaceutics12090842